Microbiomes are composed of many different species of microorganisms, and each microbiome (both intra- and inter-person) can be vastly different from another. Thus, being able to determine what an individual’s microbiome contents are is very important. To do this, the genetic code of the microbes can be sequenced. From a sample, scientists can extract the genetic code and then sequence it to know what the DNA code is. Typically, when researchers want a microbiome sample sequenced, they will pay a third party company to sequence the sample and receive electronic files with the DNA code from the sample. There are different ways to sequence a microbiomic sample: read below to learn more about 16S rRNA sequencing, shotgun metagenomics, and metatranscriptome sequencing. Most of the time, researchers will choose one method over another either because of their budget or depending on the goal of the study, but there are pros and cons to each approach.

The 16s rRNA gene is the most common gene sequenced to identify bacteria and other microorganisms. There are other genes that are in microbes, but the 16s rRNA gene is considered the most conserved throughout species, meaning it is one of the most reliable to identify the parent organism. The 16s rRNA gene is made of nine hypervariable regions which are unique to particular species. When performing 16s rRNA sequencing, two hypervariable regions are usually chosen to identify the species (see Image 2). The hypervariable regions chosen are given molecular barcodes so computational methods can later detect possible errors and identify sequences easily. The sequencing method uses polymerase chain reaction (PCR) [a] to amplify the selected range of DNA in the sample to a detectable level. Typically, next generation sequencing (NGS), a modern and rapid method for DNA/RNA analysis, is used to sequence the samples. Computational methods are then used against a reference database of known species’ genomes to determine the contents of the sample. Finally, conclusions can be made from 16S-sequenced data (see Image 3) drawing conclusions in a research study.
The shotgun metagenomic sequencing method sequences all of the DNA in the sample, then attempts to assign specific parts of the sequence to particular species from a known database. Compared to trying to assign a region from a species from a relatively small portion of the genome in 16S, metagenomic sequencing considers the whole genome. Scientists do this by using NGS to splice the samples’ genome into readable lengths and associate the fragments to a reference genome (see Image 3). This method comes with pros and cons. On the plus side, it tends to be more reliable and accurate. But on the other hand, it is significantly more expensive. Depending on the study done, researchers may opt for one over the other because of price. Though, the cost of shotgun metagenomic sequencing has been decreasing over time, which may cause an increase in popularity.
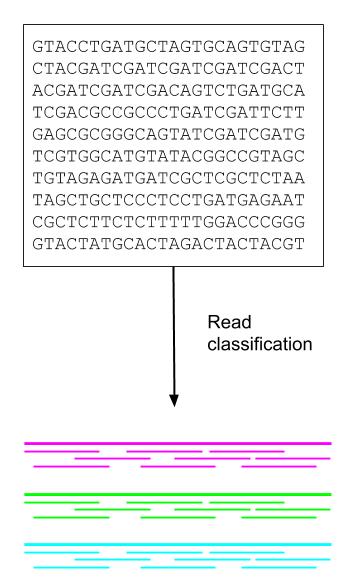
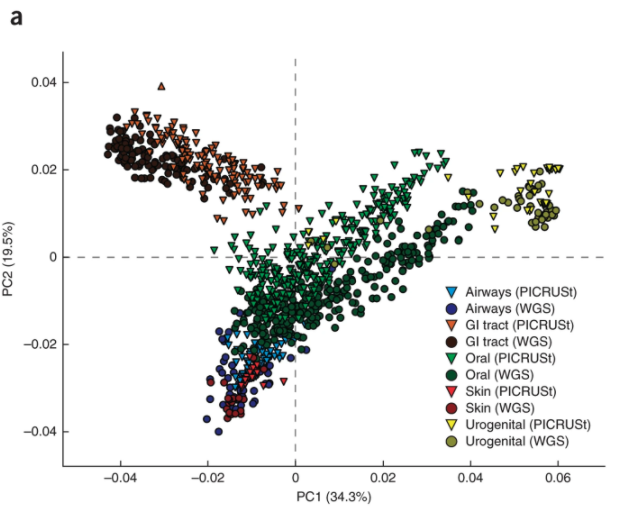
Image 4: DNA fragments (short pieces) are compared against the reference genome (solid line) during read classification to indentity sample contents.
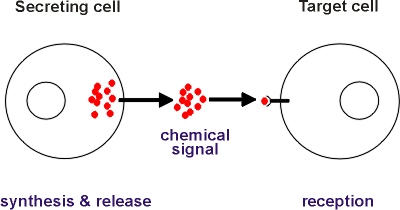
Image 5: Chemical signaling is a communication method that cells and microorganism can use to interact with one another.
This type of sequencing is foundationally different from both 16S and shotgun metagenomics, which measure the composition of the microbiome in a taxonomic- and census-style. Metatranscriptome sequencing, however, aims to analyze the microbiome sample for the organisms that are active. For background, microorganisms in the gut microbiome have a connection and interact with the host organism with chemical signals (see Image 5). Another way that the microorganisms are active is by producing proteins. To do this, they need to go through a process of protein synthesis – one step in which is to produce RNA. Metatranscriptomic sequencing, rather than measuring the DNA, measures this RNA to identify the active microorganisms. This method of sequencing is relatively new, but opens a lot of doors to learning more about microbiomes. A company called Viome specializes in this type of sequencing.
Computational power is crucial to advancement in the field of microbiomes; samples are useless if researchers have no way to tell what is in them. One of the most limiting factors in modern computational methods of microbiomic (and general genomic) analysis is the inability to process the sheer amount of data that is available. For example, the file for the genome of escherichia coli [1] [b] more commonly known as e. coli, is over 66,000 lines with 70 nucleotides-letters in each line (see Image 6), and an average microbiome has many trillions of microbes in it. [2] Though a single sample will not have every single microorganism in it, the amount of data is extremely large; there must be significant computational power to sift through all of the data and analyze it. This is part of the reason 16S rRNA sequencing is cheaper than shotgun metagenomic sequencing, since the former only does analysis on a particular gene for each organism and the latter sequences the entire genome.
There are bioinformatic [c] pipelines [d] that are free to download like QIIME (pronounced chime) and Mothur (see Images 7 and 8), and each source has its own unique procedure. Once the sequences are acquired from samples, the computational workflow for analyzing a microbiome is different for each of 16S, shotgun metagenomics, metatranscriptomics. Whichever bioinformatic tool is used has a specific pipeline and series of steps to demultiplex data [e] and align it using dynamic programming [g]. For 16S sequencing, the hypervariable regions are cross-referenced to a database of known species to determine the contents of the sample. The computer program used needs to be powerful enough to search for near-matches, not just identical matches. Because it is unlikely that the reference gene sequences will be exactly the same as the one in the sample. This is most probable to happen either because the gene of the sequenced organism itself had a naturally occurring mutation [f] or there was an error during the sequencing process – it is widely accepted that sequencing is >97% accurate. [3] Since a difference between the reference and unknown sequence is almost guaranteed, it is important that the computer program compensate for that. On the other hand, if the program is too lax in how many differences it allows, it will falsely classify certain 16S genes as a species. There are algorithms that attempt to diminish the computational power needed to do this analysis called dynamic programming [g]. For metagenomic and metatranscriptomic sequencing, similar methods are used but on a different scale. The former analyzes the entire genome of the microorganisms and the latter analyzes the present RNA in a sample. While these methods have made these processes significantly less time and power consuming than brute-force methods, large datasets continue to pose a challenge for microbiome analysis.

Image 6: The beginning of e coli's genome in the form of a .fasta file.

Image 7: Qiime logo.

Image 8: Mothur logo.
So what? Find out why microbiomes are important here.